
The Oncogenic Enigma of KMT2A Rearrangements in Acute Leukemia
Acute leukemias characterized by rearrangements of the Lysine-specific N-methyltransferase 2A (KMT2A) gene represent some of the most aggressive and clinically challenging hematologic malignancies. Found in a significant subset of Acute Myeloid Leukemia (AML) and Acute Lymphoblastic Leukemia (ALL), these cancers are notorious for their resistance to conventional chemotherapy and high rates of relapse, leading to dismal patient outcomes. Understanding the molecular underpinnings of KMT2A-rearranged (KMT2A-r) leukemia is paramount for developing the targeted, effective therapies needed to change this prognosis. This requires a deep dive into the normal function of the KMT2A protein, the catastrophic consequences of its genetic rearrangement, and the diverse landscape of its fusion partners.
Normal Function of the KMT2A Protein
The KMT2A gene, also known as Mixed-Lineage Leukemia (MLL), is located on chromosome 11q23 and encodes a large, multi-domain protein that is a master regulator of gene expression. Its primary function is that of a transcriptional coactivator. A key component of its activity resides in its C-terminal SET domain, which possesses histone H3 lysine 4 (H3K4) methyltransferase activity. H3K4 methylation is a powerful epigenetic modification associated with active gene promoters, essentially flagging genes to be turned on by the cell’s transcriptional machinery.
In its normal state, the KMT2A protein is crucial for fundamental biological processes, including embryonic development and, most critically, hematopoiesis, the formation of blood cells. It achieves this by regulating the expression of key developmental genes, most notably the HOX gene clusters (e.g., HOXA9, HOXA7) and their essential cofactors like MEIS1 and PBX3. These genes orchestrate the delicate balance between hematopoietic stem cell self-renewal and differentiation. For proper function, the full-length KMT2A protein undergoes proteolytic cleavage by the enzyme Taspase-1 into two fragments, an N-terminal portion (KMT2A-N) and a C-terminal portion (KMT2A-C), which then reassociate to form the active KMT2A multiprotein complex. This processing is essential for its correct localization and regulatory activity.
The Molecular Origin of KMT2A Fusion Proteins
KMT2A rearrangements arise through chromosomal breaks spanning approximately 8.3 kb across introns 9 to 11 (transcript-dependent). A smaller, distal breakpoint region has also been described in a subset of cases near intron 21. Because exon and intron boundaries vary slightly between transcript annotations and across studies, this region is typically referenced as an extended hotspot rather than a fixed coordinate window. Importantly, regardless of the exact breakpoint position, all clinically relevant fusions retain the N-terminal chromatin-tethering modules of KMT2A, which support menin-dependent genomic anchoring and drive the downstream leukemogenic transcriptional program.
Most KMT2A fusion proteins preserve the N-terminal DNA- and chromatin-binding modules while losing the C-terminal SET domain that normally provides intrinsic H3K4 methyltransferase activity. Rather than abolishing interaction with the native C-terminal region, these chimeric proteins acquire oncogenic function by recruiting alternative transcriptional and epigenetic machinery, including DOT1L-associated complexes, and by leveraging menin-mediated chromatin tethering. The combined effect is an aberrant activation of HOX and MEIS1 that sustains leukemic stemness and defines the therapeutic vulnerability to menin inhibition.
Key Fusion Partners and Their Prognostic Implications
The KMT2A gene is notoriously promiscuous, with over 80 different fusion partners identified to date. While many are rare, a handful of recurrent partners are responsible for the majority of clinical cases. The identity of this fusion partner is a critical determinant of the disease’s biology and, consequently, the patient’s prognosis.
In AML, the most frequent partners include MLLT3 (also known as AF9), MLLT10 (AF10), MLLT1 (ENL), and ELL. In ALL, particularly in infants, the most common partners are AFF1, MLLT1, and MLLT3. The specific fusion partner dictates more than just the name of the translocation; it appears to program the entire biological behavior of the leukemia. For instance, in pediatric AML, patients with the KMT2A::MLLT3 fusion generally have a more favorable prognosis compared to those with other fusions like KMT2A::MLLT10 or KMT2A::AFDN. Conversely, the KMT2A::AFF1 fusion in ALL is associated with a particularly aggressive disease course and very poor outcomes. In addition to translocations, another pathogenic alteration is the KMT2A partial tandem duplication (PTD), an internal rearrangement of the gene that also drives leukemia.
Why KMT2A-r Leukemias Portend a Poor Prognosis
Regardless of the specific fusion partner, KMT2A-r leukemias are consistently classified as high-risk diseases. They are present in up to 10% of all acute leukemias and are accounting for approximately 75-80% of leukemia cases in infants.
The clinical presentation is often aggressive, characterized by hyperleukocytosis (an extremely high white blood cell count) and a high incidence of central nervous system involvement. Patients frequently exhibit primary resistance to standard induction chemotherapy, and even when remission is achieved, it is often shallow and short-lived, with a high rate of early relapse. The inability to achieve a deep, minimal residual disease (MRD)-negative remission is a key driver of this poor prognosis. Consequently, long-term overall survival rates for patients with KMT2A-r leukemia remain unacceptably low, often 25-30% in infants ALL to 35-50% in adults AML. This dire clinical reality has fueled an urgent search for novel therapeutic strategies that can specifically target the core molecular machinery driving KMT2A-r leukemias.
Dr. Morgan Drucker, a Pediatric Hematology/Oncology Fellow at Cincinnati Children’s Hospital, presents single-cell multiomic datasets generated by her group to dissect clonal architecture in acute leukemias. Her analysis centers on two biologically distinct cohorts, adult patients with NPM1-mutated AML and pediatric patients harboring KMT2A-rearranged leukemias. Despite their differing clinical contexts, both disease subsets are driven by genomic lesions (NPM1 mutations or KMT2A fusions) that depend on the functional interaction between menin and the KMT2A complex, making them highly relevant for menin-targeted therapeutic strategies.
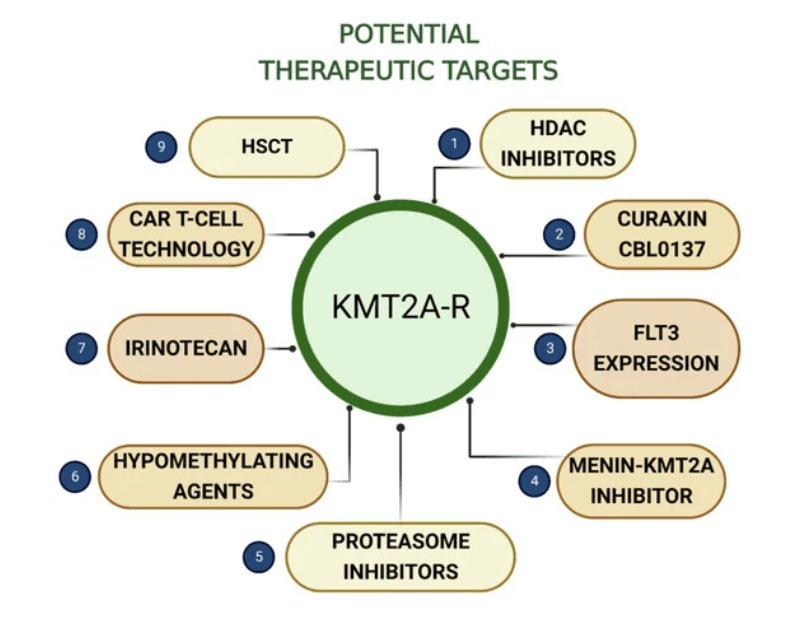
Targeting the Menin-KMT2A Complex: A New Leukemia Therapy Approach
For decades, the treatment of KMT2A-r leukemias relied on chemotherapy, an approach that failed to address the specific molecular driver of the disease. The breakthrough came with the discovery of a critical dependency: the leukemogenic activity of KMT2A fusion proteins depends on their interaction with the nuclear scaffold protein menin. This discovery unveiled a key therapeutic vulnerability for a new class of targeted drugs, menin inhibitors, that are shaping how we treat KMT2A-r leukemias.
The Role of Menin in KMT2A-r Leukemogenesis
Menin is a protein encoded by the MEN1 gene, a tumor suppressor in endocrine tissues but an essential oncogenic cofactor in the context of KMT2A-r leukemia. Researchers discovered that menin binds directly to a highly conserved region within the N-terminus of the KMT2A protein, the very region that is retained in all KMT2A fusion proteins. This physical interaction acts as an anchor, tethering the entire KMT2A fusion complex to chromatin at the promoter regions of its target genes, such as HOX and MEIS1.
In preclinical models, genetically deleting menin or introducing mutations that block its binding to KMT2A completely abrogated the ability of the fusion protein to drive leukemia. This established the menin-KMT2A interaction as the “Achilles’ heel” of KMT2A-r leukemias. Further studies also revealed a convergence of oncogenic pathways. A distinct subtype of AML, characterized by mutations in the Nucleophosmin 1 (NPM1) gene, also exhibits a similar gene expression signature, with high levels of HOX and MEIS1. It was found that this subtype, despite lacking a KMT2A rearrangement, is also dependent on the interaction between menin and the wild-type KMT2A protein to drive its leukemogenic program. This expanded the potential clinical utility of targeting the menin-KMT2A complex to a much larger group of AML patients.
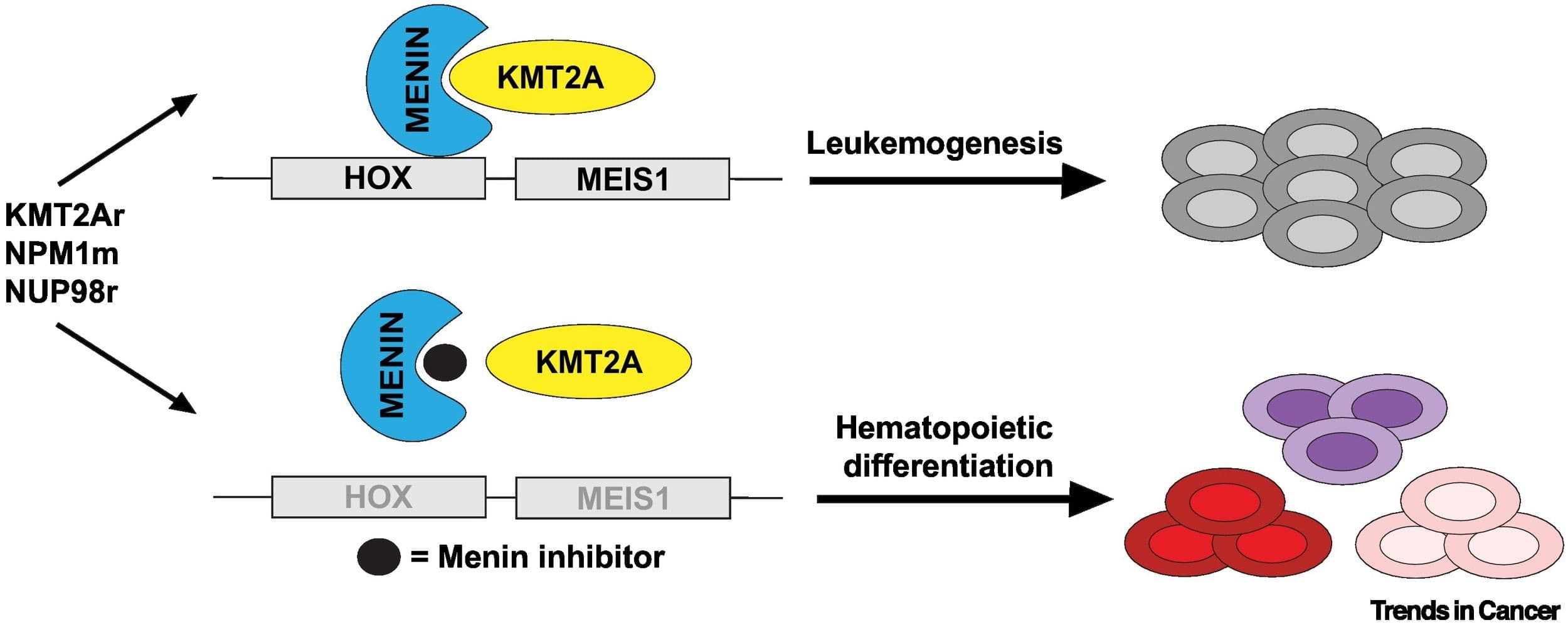
How Menin Inhibitors Dismantle the Oncogenic Complex
Menin inhibitors are a novel class of oral, small-molecule drugs designed to function as protein-protein interaction (PPI) disruptors. Unlike traditional enzyme inhibitors that block a catalytic site, these drugs are engineered to physically obstruct the interface between two proteins. They fit precisely into a binding pocket on the surface of the menin protein, the same pocket that the KMT2A protein normally occupies.
Menin inhibitors bind the menin pocket and block its interaction with KMT2A. Disruption of this interaction displaces the oncogenic complex from HOX/MEIS1 loci, downregulates the HOX-driven leukemogenic program, relieves the differentiation block, and induces differentiation or cell death in preclinical models and validated by early clinical responses. While this represents a key therapeutic vulnerability for KMT2A-r leukemias, resistance mechanisms have been observed and are an active area of investigation.
In clinical trials of menin inhibitors, differentiation syndrome has emerged as an on-target pharmacodynamic effect, reflecting the rapid release of the leukemic differentiation block and triggering a systemic immune and inflammatory response. While the syndrome can be severe and requires prompt management with corticosteroids and supportive care, its occurrence strongly suggests that the drug is engaging its target and driving the intended biological effect. It is important to note, however, that differentiation syndrome is not universal and many patients respond to therapy without developing this syndrome.
Clinical Development and Trial Data of Leading Menin Inhibitors
Several menin inhibitors have rapidly advanced through clinical development, demonstrating significant efficacy and culminating in the first regulatory approval within this class. These small-molecule therapeutics, including revumenib and ziftomenib, act as mechanism-based inhibitors that selectively disrupt the menin-KMT2A interaction which is a critical dependency in KMT2A-rearranged and NPM1-mutated acute leukemias. By blocking this protein–protein interface, menin inhibitors dismantle the oncogenic transcriptional complex that sustains HOXA9 and MEIS1 overexpression, leading to myeloid differentiation, cell-cycle arrest, and subsequent apoptosis.
Revumenib (Revuforj®), developed by Syndax Pharmaceuticals, was the first menin inhibitor to gain FDA approval. On November 15, 2024, the FDA approved Revuforj for adult and pediatric patients (≥1 year) with relapsed or refractory acute leukemia harboring a KMT2A translocation. The approval was based on the pivotal AUGMENT-101 trial, which reported a complete remission (CR) or complete remission with partial hematologic recovery (CRh) rate of about 21–23%. Among those achieving CR/CRh with available MRD data, roughly 70% were MRD-negative, and the median duration of CR/CRh was 6.4 months (95% CI, 3.4–not reached). This enables further treatment to potentially curative hematopoietic stem cell transplantation (HSCT).
Ziftomenib, developed by Kura Oncology in partnership with Kyowa Kirin, has shown similarly impressive results, particularly in patients with R/R NPM1-mutant AML(R/R NPM1-m AML). In the KOMET-001 trial, ziftomenib monotherapy achieved a CR/CRh rate of approximately 23-25%. Based on these data, it received Breakthrough Therapy Designation from the FDA for R/R NPM1-m AML patient population. Kura is now advancing ziftomenib into large-scale Phase 3 trials (KOMET-017) evaluating its use in combination with standard-of-care chemotherapy in the frontline setting, aiming to establish menin inhibition as a core component of initial therapy for both KMT2A-r and NPM1-mutant AML.
What Data is Most Important for Developing Safe and Effective Therapies for KMT2A-r Leukemias?
The advent of targeted therapies like menin inhibitors has shaped how we approach treatment for KMT2A-rearranged leukemias, but their optimal use hinges on collecting the right data to fully characterize therapeutic response. Acute leukemias are not monolithic entities but dynamic ecosystems of genetically and epigenetically diverse clones, a phenomenon known as intratumoral heterogeneity. To effectively diagnose, treat, and monitor these diseases, it is no longer sufficient to measure the “bulk average” characteristics of a cell population. We must deconstruct this complexity at the single-cell level, integrating DNA, RNA, and protein insights to understand mechanisms of action, track minimal residual disease (MRD), and uncover emergent resistance pathways.
Limitations of Bulk Sequencing in Heterogeneous Cancers
For years, the molecular characterization of cancer has relied on bulk sequencing methods. These techniques analyze DNA or RNA extracted from millions of cells simultaneously, providing a powerful but fundamentally averaged view of the tumor’s genetic and transcript makeup. While bulk sequencing can identify the dominant mutations and gene expression patterns, it inherently masks the underlying cellular diversity that is distinct to cancer cell populations.
In a disease like KMT2A-r leukemia, this averaging can have critical clinical consequences. A small, pre-existing subclone harboring a resistance mutation may be completely invisible in the bulk data, drowned out by the signal from the dominant population. This means the clonal architecture (how different subpopulations of cancer cells are related to each other) cannot be resolved. This limitation means that clinicians may be making treatment decisions based on an incomplete and potentially misleading picture of the patient’s disease. Additionally, scientists developing new therapies could be targeting the wrong clones that are masking the true cells that drive disease progression and relapse.
A Multi-Layered View: The Principles and Power of Single-Cell Multi-omics
Single-cell multiomics overcome bulk averaging limitations by enabling the simultaneous measurement of multiple layers of molecular information such as DNA, RNA, and proteins from the same individual cell. This capability allows for the direct and unambiguous linking of a cell’s genotype to its phenotype. For example, one can determine that a specific leukemic cell with a KMT2A-MLLT1 fusion and a co-occurring NRAS mutation expresses a particular set of genes and displays a specific profile of proteins on its surface.
Using single-cell multiomics assays in therapy development for diseases like KMT2A-r leukemia shifts analysis from an averaged overview to a detailed, cell-by-cell understanding of the sample composition. By examining thousands of individual cells within a patient sample, single-cell multiomic defines the genetic, transcriptomic, and proteomic features of each cell. This data can reveal how different clones and lineages are organized, how they interact within the tumor microenvironment, and how specific cell states contribute to disease progression or therapeutic response. The result is a comprehensive view of cancer’s heterogeneity and the biological processes driving it.
Applications in Hematologic Malignancies: Mapping Clonal Evolution, Resistance, and MRD
The clinical utility of single-cell multiomics in KMT2A-r leukemia could span the entire patient journey, from diagnosis to relapse.
- Diagnosis and Risk Stratification: At diagnosis, the technology can precisely identify not only the specific KMT2A fusion partner but also the full spectrum of co-occurring mutations within each distinct leukemic clone. This detailed clonal map provides a much more accurate basis for risk stratification and initial treatment planning.
- Therapy Selection: By measuring protein expression and signaling pathway activity at the single-cell level, researchers can confirm that the molecular target of a small-molecule inhibitor is expressed and active within cancer cells. This helps ensure that the drug engages its intended target and distinguishes tumor-specific signaling from that of surrounding healthy cells.
- Monitoring minimal residual disease (MRD): Perhaps the most powerful application is in the detection and characterization of MRD. Standard MRD techniques can quantify the number of residual leukemic cells, but single-cell multiomics MRD assays for AML can reveal their molecular identity. It can determine which specific clone survived initial therapy, whether it has acquired new mutations, and what its transcriptional and proteomic vulnerabilities are. This information is critical for designing rational therapeutic strategies to eradicate these persistent cells and prevent relapse.
How Can Single-Cell Multiomic Analysis Help The Development of New Drugs Menin Inhibitors For KMT2A-r Leukemia?
To illustrate the power of single-cell multiomic in a practical context, a single-cell multiomic assay was developed to be applied to four distinct cell lines using the Mission Bio Tapestri platform. This experiment serves as a compelling demonstration of how an integrated, five-layer molecular analysis can precisely identify and distinguish cell populations with features highly relevant to clinical leukemia.
Profiling Leukemia Using Single-Cell Multiomics
To demonstrate the utility of single-cell multiomics in characterizing leukemia, a custom, fit-for-purpose assay was developed to profile four cell lines representing distinct biological states or leukemia-associated mutations:
- GM24385: well-characterized lymphoblastoid line used as a control for benchmarking variant detection and well known to be part of the genome in a bottle project. Although germline variants are present, it serves as a non-malignant control for somatic variant analyses.
- Kasumi-6: An AML cell line characterized by dysregulated myeloid differentiation
- OCI-AML3: A model of NPM1- and DNMT3A-mutant AML
- OCI-AML4: Less extensively characterized AML model with distinct transcriptional and mutational features relative to OCI-AML3.
Together, these models represent diverse genetic and phenotypic states found across leukemia. Using a single-cell multiomic assay to resolve these distinct transcriptional, proteomic, and mutational profiles of each cell line demonstrates the assay’s analytical capabilities in clinical settings and diagnostics for investigating KMT2A-rearrangement and menin inhibitor responses.
Layer-by-Layer Analysis
The assay simultaneously generated information across five molecular layers, producing a high-resolution “portrait” of each cell population.
Layer 1: DNA variants (SNVs/Indels) Single-cell DNA sequencing identified the defining pathogenic mutations characteristic of each AML model while confirming the wild-type status of the normal wild type line.
- OCI-AML3 exhibited single-cell validation of its canonical NPM1 and DNMT3A mutations consistent with its published genotype. NRAS mutation was also detected but with uncertain significance.
- Kasumi-6 and OCI-AML4 displayed distinct patterns of leukemia-associated variants, enabling a clear clonal separation from each other and from OCI-AML3.
- GM24385 displayed no pathogenic variants for the pathogenic targets evaluated.
Layer 2: Copy Number Variation (CNV) in KMT2A Gene-level CNV profiling revealed the ability of single-cell CNV analysis to detect KMT2A structural abnormalities that may influence mini dependency or fusion biology.
- Kasumi-6 displayed an elevated apparent copy number across exons within the PTD region, consistent with local duplication from KMT2A-PTD rather than whole-gene amplification.
- OCI-AML3 and GM24385 maintained relatively diploid copy number states across the gene.
- OCI-AML4 surprisingly displayed a slight elevation of copy number for KMT2A.
Layer 3: KMT2A Fusion Transcripts RNA-based fusion calling identified KMT2A-PTD and KMT2A-MLLT1 transcripts specific to AML where each line displayed a distinct fusion profile. These results highlight how fusion detection at single-cell resolution can discriminate between closely related AML subtypes.
- Kasumi-6 demonstrated expression of KMT2A-PTD consistent with published characterization of its complex KMT2A architecture.
- OCI-AML4 demonstrated KMT2A-MLLT1 fusion transcript reads in this assay, although this fusion is not a widely published feature of OCI-AML4. The signal likely reflects either a subclone, low-level rearrangement, or assay-specific detection.
- OCI-AML3 and GM24385 showed no KMT2A fusions, as expected.
Layer 4: Gene Expression (HOXA/MEIS1) Transcriptional profiling revealed differential activation of canonical KMT2A-rearranged leukemic programs demonstrating how single-cell targeted RNA profiling can quantify downstream pathways activation relevant to menin inhibitor response.
- Kasumi-6 and OCI-AML4 displayed heightened expression of HOXA9, MEIS1, and related homeobox partners, consistent with KMT2A-driven transcriptional dysregulation.
- OCI-AML3 exhibited a distinct expression pattern reflective of NPM1+/DNMT3A+ AML biology.
- GM24385 showed minimal expression of these leukemogenic transcription factors.
Layer 5: Cell Surface Protein Expression (Immunophenotype) The linkage between genotype, transcriptome, and immunophenotype mirror the diagnostic framework used in clinical hematopathology and MRD assessments. Therefore protein profiling is essential to link each genotype to its corresponding immunophenotype.
- GM24385 primarily expresses B-cell markers (CD19, CD22), though low-level expression of myeloid antigens in the figure likely reflects assay background or nonspecific signal rather than true biological expression.
- Kasumi-6, OCI-AML3, and OCI-AML4 each expressed myeloid-restricted markers such as CD13 and CD33, with each line exhibiting a unique combination of antigens.
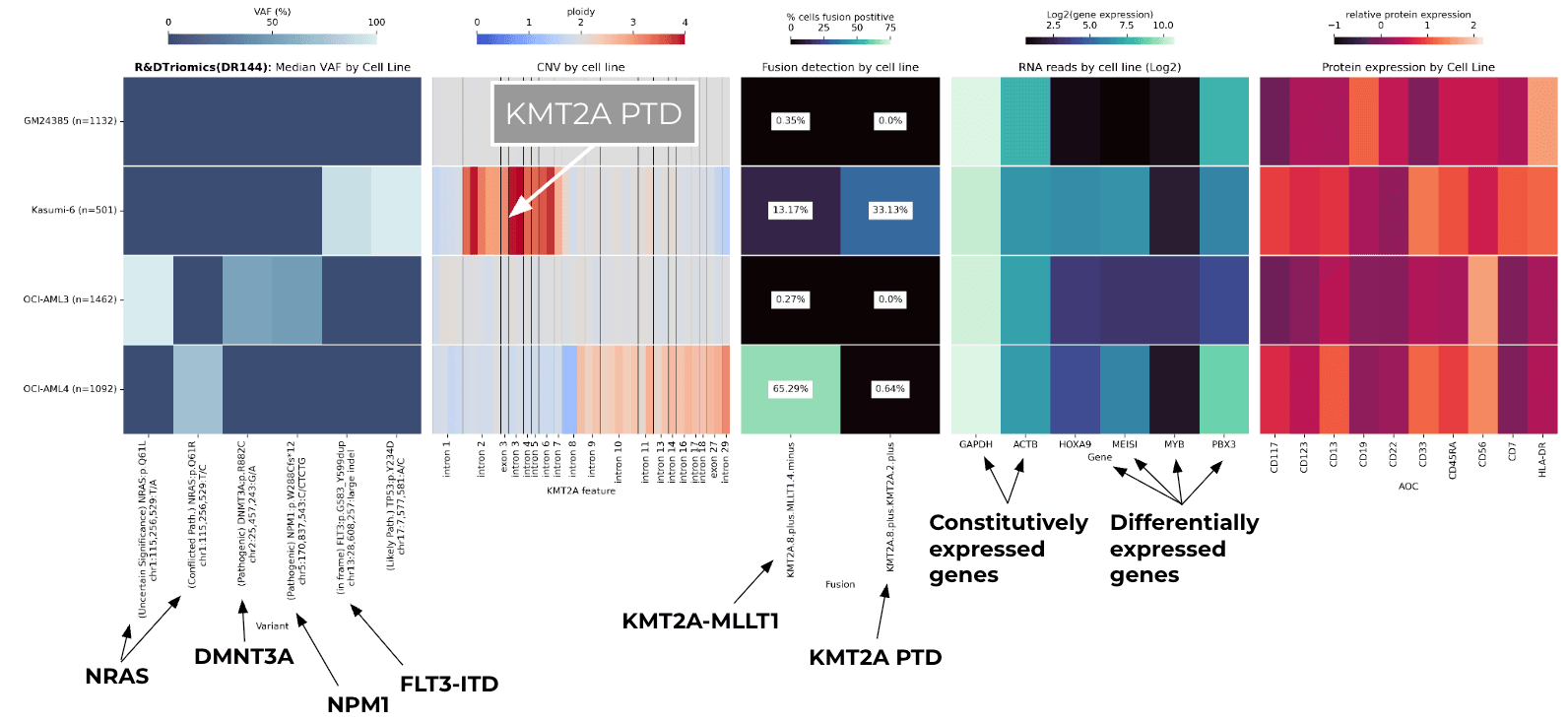
Building an Integrated, High-Resolution Molecular Portrait for KMT2A Rearrangement AML
By integrating these five layers of data, a comprehensive molecular “portrait” of each cell line emerges. The power of the single-cell multiomic approach lies in its ability to synthesize these disparate data types into a single, coherent narrative for each cellular population. The integration of SNVs, CNVs, fusion status, transcriptional programs, and protein phenotypes allows each cell line to be unambiguously distinguished along multiple orthogonal axes revealing:
- How genetically similar AMLs diverge in transcriptional output
- How KMT2A structural variation manifests differently across models
- How fusion biology correlates with HOXA/MEIS1 activation
- How immunophenotype maps directly onto underlying genotypes
This case study illustrates how single-cell multiomic profiling can concurrently resolve SNVs, CNVs, fusion transcripts, transcriptional activity, and immunophenotypic states within individual cells. By precisely identifying structurally complex KMT2A lesions such as partial tandem duplications and focal amplifications, alongside co-occurring signaling mutations like FLT3 and NRAS, this approach exposes the full spectrum of menin-dependent and menin-independent subclones that shape disease biology in KMT2A-rearranged AML. These insights enhance MRD interpretation for patients treated with menin inhibitors and support more informed therapeutic decisions, including escalation, combination strategies, and monitoring of high-risk clonal trajectories.
Using Single-Cell Multiomics in Clinical Practice and Drug Discovery to Treat Leukemia
High-resolution diagnostic technologies that use single-cell multiomics are influencing how KMT2A-rearranged leukemias are studied and treated. By understanding clonal heterogeneity and linking genetic mutations to transcriptional readouts and proteomic phenotypes, these approaches provide valuable single-cell insights into disease mechanisms and therapeutic response. This type of data has the ability to support the development and refinement of targeted therapies, such as menin inhibitors, and enables patient stratification based on measurable molecular attributes.
Smarter Drug Development and Patient Stratification for KMT2A-r Leukemia
For clinical utility, single-cell multiomics could help refine patient selection for targeted KMT2A-r leukemia therapies. By providing a complete molecular profile at diagnosis, these single-cell multiomics assays can more accurately identify the patients most likely to benefit from menin inhibitors, specifically those whose leukemic clones are driven by KMT2A rearrangements or NPM1 mutations. Equally important, they can identify patients whose disease is driven by co-occurring pathways, such as RAS mutations. Rather than simply excluding these patients, this insight highlights the need for upfront combination strategies to prevent the rapid emergence of resistance often seen with monotherapy.
Furthermore, by mapping all co-occurring mutations at the single-cell level, it becomes possible to identify mechanisms with de novo resistance. For example, the presence of a subclone with a KRAS mutation alongside a KMT2A fusion could signal a lower likelihood of a deep response to menin inhibitor monotherapy, suggesting the need for an upfront combination strategy. Additionally, single-cell multiomics can provide a powerful platform for understanding acquired resistance. If a patient relapses on a menin inhibitor, single-cell multiomic analysis of the relapse sample can determine if a subclone has emerged with a secondary mutation in the MEN1 gene, which would render the drug ineffective and a new treatment strategy would need to be administered.
Clinical Diagnostic For MRD Detection and Dynamic Monitoring
Current standard-of-care minimal residual disease (MRD) testing should provide a quantitative count of surviving cancer cells. However, the future of MRD monitoring lies not just in detecting that residual cells exist, but in comprehensively characterizing them. Single-cell multiomic analysis of MRD for AML can reveal the complete genotype, transcriptome, and proteome of rare, therapy-resistant cells. This deeper molecular characterization provides actionable insights that bulk average assessments miss, avoiding blind spots that could compromise patient safety.
This deeper molecular characterization of each cell provides actionable insights that bulk average assessments miss which could compromise the safety and wellbeing of a patient. For instance, single-cell multiomics could reveal that the surviving clone has upregulated an anti-apoptotic pathway or a specific cell surface receptor. This information can then be used to design a rational, targeted consolidation or maintenance therapy aimed specifically at eradicating that particular clone’s vulnerabilities, thereby preventing a clinical relapse. This moves MRD from a prognostic biomarker into a predictive tool for guiding subsequent therapeutic interventions for diseases like KMT2A-rearrange AML.
Uncovering New AML Targets and Overcoming Resistance Beyond KMT2A-r
The application of single-cell multiomics to assess a patient’s treatment journey from diagnosis to drug administered and at relapse, creates a clinical utility that enhances patient care. By comparing the molecular profiles of sensitive and resistant cells from the same patient, researchers can identify the novel biological pathways that cancer cells activate to evade therapy. This approach dramatically accelerates the drug discovery cycle by moving directly from a clinical observation of resistance to a set of testable hypotheses about its underlying mechanism. The pathways identified in resistant clones represent a rich source of next-generation therapeutic targets, paving the way for new drugs and combination strategies designed to overcome resistance before it becomes clinically apparent.
The Clinical Utility of Single-Cell Multiomics as a Precision Diagnostic for KMT2A-r Leukemia and Other Hematology-Oncology Diseases
The application of single-cell multiomics to a patient’s journey, from diagnosis through treatment and at relapse, offers powerful clinical utility that directly enhances patient care. By precisely comparing the molecular profiles of sensitive and resistant cells from the same individual, researchers can pinpoint the biological pathways cancer cells activate to evade therapy. This capability is particularly important for patients with KMT2A-rearranged leukemias, long considered among the most challenging hematologic malignancies.
The development of menin inhibitors has provided a potent, targeted weapon against KMT2A-rearranged leukemias. However, the full potential of these menin inhibitors and other precision therapies can only be realized when guided by an equally precise diagnostic. Single-cell multiomics provides the necessary single-cell resolution to understand the heterogeneity and evolutionary dynamics of leukemia. Combining powerful targeted therapeutics with single-cell diagnostic platforms brings a new era in hematology-oncology treatment. In this new era, single-cell multiomic diagnostics allow treatment to be dynamically adapted in real-time to the evolution of the disease, promising to finally change the prognosis for patients battling these aggressive cancers and bringing a cure within reach.
To see more data on how single-cell multiomics helped a pharmaceutical company develop a menin inhibitor for KMT2A-r Leukemia, contact us to learn more.
References
Rao R, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–346.
Kühn MW, Song E, et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov. 2016;6(10):1166-1181.
Richard-Carpentier G, Kantarjian HM, et al. Outcomes of acute lymphoblastic leukemia with KMT2A (MLL) rearrangement: the MD Anderson experience. Blood Adv. 2021;5(23):5415–5419.
Górecki M, Kozioł I, et al. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines. 2023;11(3):821.
Mrózek K. Prognostic importance of the fusion partners and measurable residual disease in patients with acute myeloid leukemia who harbor 11q23/KMT2A alterations. Transl Pediatr. 2023;12(10):1920-1925.
Candoni A, Coppola G. A 2024 Update on Menin Inhibitors. A New Class of Target Agents against KMT2A-Rearranged and NPM1-Mutated Acute Myeloid Leukemia. Hematol Rep. 2024;16(2):244-254.
Guarnera L, D’Addona M, et al. KMT2A Rearrangements in Leukemias: Molecular Aspects and Therapeutic Perspectives. Int J Mol Sci. 2024;25(16):9023.
Ogino J, Dou Y. Histone methyltransferase KMT2A: Developmental regulation to oncogenic transformation. J Biol Chem. 2024;300(10):107791.
Zehtabcheh S, Soleimani Samarkhazan H, et al. Insights into KMT2A rearrangements in acute myeloid leukemia: from molecular characteristics to targeted therapies. Biomark Res. 2025;13(1):73.



